Data presentazione
28 settembre 2016
Responsabile Scientifico
Prof Francesco Passamonti
Dipartimento di Medicina Clinica e Sperimentale Università degli Studi dell’Insubria, Varese
Riassunto del progetto
Le neoplasie meloproliferative croniche (NMC), quali la policitemia vera (PV), la trombocitemia essenziale (TE), la mielofibrosi primaria (MFP) e secondaria (MFS) sono malattie rare. L’attuale cura di queste malattie consta di un approccio osservazionale delle condizioni a basso rischio e di una terapia citoriduttiva se la malattia si presenta ad alto rischio vascolare. Ruxolitinib (inibitore di JAK2) e trapianto di cellule staminali emopoietiche sono riservate a condizioni di malattia più avanzata. Queste neoplasie riconoscono una base molecolare in parte comune, legata alla presenza di mutazioni dei geni JAK2, CALR, MPL determinanti il fenotipo clinico. Queste mutazioni sono presenti nel 90% dei pazienti con NMC. Circa il 10% dei pazienti con TE e MF non presenta queste mutazioni e sono riconosciuti come tripli negativi. Questo progetto di medicina personalizzata si pone come obiettivo lo studio molecolare di queste malattie attraverso metodiche di targeted sequencing. Con metodiche di next generation sequencing (NGS) si definirà la carta d’identità molecolare dei pazienti con NMC. Integrando dati clinici e molecolari, si svilupperanno modelli prognostici in grado di predire in modo accurato la sopravvivenza dei pazienti e la risposta ai nuovi farmaci o al trapianto di cellule staminali emopoietiche. Il network di patologia nel quale si svilupperà lo studio è la Rete Ematologia Lombarda (REL). Il progetto porterà a una personalizzazione della cura per il singolo paziente con NMC.
Background
Le neoplasie mieloidi sono malattie rare che comprendono le leucemie acute (LA), le sindromi mielodisplastiche (SMD) e le neoplasie meloproliferative croniche (NMC), quali la policitemia vera (PV), trombocitemia essenziale (TE) e la mielofibrosi (MF) primaria (MFP) e secondaria (MFS).[1,2] Queste ultime condizioni hanno un naturale rischio di complicanze vascolari, quali trombosi ed emorragie, e di evoluzione clonale in LA e MFS. La loro incidenza annua è 1-2 x 100.000 individui. Le conoscenze patogenetiche di queste malattie sono in continuo sviluppo. Ad esempio, nelle NMC sono state indentificate mutazioni del DNA che determinano il fenotipo clinico della malattia (phenotypic driver mutations), quali JAK2, MPL e CALR. Oltre a queste mutazioni sono presenti altre mutazioni con un ruolo addizionale quali ASXL1, EZH2, CBL, IDH1/IDH2, TP53, SRSF2 di cui alcune con ruolo centrale nell’evoluzione clonale (clonal driver mutations). Alcune di queste mutazioni sono presenti anche nelle SMD e nelle LA de novo. La definizione delle basi molecolari di queste malattie ha migliorato la conoscenza di queste malattie favorendone la definizione prognostica (modello DIPSS per la mielofibrosi,[3] modello IPSET per la trombocitemia[4]), iniziando un percorso di medicina personalizzata (MP).[2] MP significa personalizzare la strategia terapeutica, cioè scegliere per ogni paziente la terapia migliore al momento migliore. Utilizzando quest’approccio Il nostro gruppo, collaborando con altri colleghi europei e americani, ha definito recentemente l’indicazione al trapianto allogenico di cellule staminali emopoietiche nella mielofibrosi.[5] L’avanzamento delle conoscenze molecolari ha portato infine alla scoperta di nuove terapie con target molecolare quali gli inibitori di JAK2. Uno tra questi farmaci, ruxolitinib, è entrato da poco a far parte delle possibilità terapeutiche della MF anche in Italia, prolungandone la sopravvivenza dei pazienti affetti.[6] Rimane ancora da identificare quali siano i pazienti che maggiormente ne beneficiano.
Obiettivi del progetto
Gli obiettivi del progetto, che si svolge in tre anni, sono:
- Generare un database delle NMC a livello di REL. Questo consentirà di raccogliere informazioni cliniche e terapeutiche sui pazienti con NMC su base multicentrica e di integrarle con i dati molecolari.
- Genotipizzare pazienti con NMC. Saranno utilizzate metodiche di targeted sequencing applicando Next Genaration Sequencing (NGS); l’ampia analisi mutazionale dei geni target consentirà l’identificazione di eventuali nuove mutazioni e la definizione della carta d’identità molecolare dei pazienti.
- Studio di nuovo percorsi di anatomia molecolare nei pazienti senza phenotype driver mutations. Saranno studiate le anomalie cromosomiche e le variazioni del numero di coppie (copy number variation, CNV) applicando l’ibridazione genomica comparativa su microarray (Comparative Genomic Hybridization – CGH). Nei pazienti con NMC senza phenotypic-driver mutations identificheremo quindi nuovi marcatori di clonalità (anomalie cromosomiche, CNV, duplicazioni/amplificazioni o delezioni).
- Sviluppare modelli prognostici integrati clinici e molecolari. Questi modelli serviranno a predire la sopravvivenza dei pazienti e la risposta alle terapie quali ruxolitinib e trapoanto di cellule staminali emopoietiche.
Centri partecipanti
Questo progetto prevede la collaborazione tra l’Unità di Ematologia del Dipartimento di Medicina Clinica e Sperimentale, Università dell’Insubria, ASST Settelaghi, Ospedale di Circolo di Varese (Direttore: Prof. Francesco Passamonti), la SSD Laboratorio Analisi, SMEL specializzato in Genetica Medica dell’ASST Settelaghi, Ospedale di Circolo, Varese (Direttore: Dott. Rosario Casalone) e altri Centri italiani o stranieri finalizzata alla realizzazione del progetto. La REL- commissione neoplasie mieloproliferative croniche – parteciperà con i suoi centri di Ematologia alla casistica.
Ipotesi
Lo studio dello stato mutazionale dei pazienti con tumori emopoietici consente di identificare le basi molecolari dei pazienti e raggrupparli per omogeneità mutazionale. L’ipotesi di questo progetto di medicina personalizzata è generare modelli prognostici integrando dati clinici e molecolari in grado di predire la sopravvivenza dei pazienti e la risposta a ruxolitinib e al trapianto di cellule staminali emopoietiche. La personalizzazione della diagnosi e della prognosi guiderà la cura più appropriata.
Disegno sperimentale
Disegno sperimentale dell’obiettivo #1.
I pazienti con diagnosi di NMC che si rivolgono all’Unità di Ematologia dell’Università degli Studi dell’Insubria, ASST Settelaghi, Ospedale di Circolo di Varese saranno inclusi nel progetto, previa firma del consenso informato. La diagnosi sarà posta in accordo alla Classificazione dell’Organizzazione Mondiale della Sanità (WHO, World Health Organization) pubblicata nel 2016, mentre la diagnosi di MFS secondo i criteri IWG-MRT (International Working Group on Myeloproliferative neoplasms Research and Treatment) pubblicati nel 2008. Il progetto ha una solida base clinica includendo 650 pazienti di cui 580 genotipizzati per le phenotype driver mutations e in follow-up presso L’Ematologia di Varese. Durante lo svolgersi del progetto ci attendiamo di raccogliere materiale clinico e genetico per un totale di 800 pazienti a Varese. Il materiale genomico sarà raccolto in accordo ai criteri standardizzati del progetto bioREL (www.rellombardia.net). La Rete Ematologica Lombarda è organizzata in Commissioni. Una tra queste è focalizzata sulle NMP e coordinata dall’Unità di Ematologia di Varese. La Commissione parteciperà al progetto condividendo il database clinico e le informazioni molecolari necessarie. L’Unità di Varese servirà i Centri REL per le analisi molecolari qualora non fattibili localmente. Allo stato attuale, è già in corso un progetto sulla ferrochelazione nella mielofibrosi che coinvolge i centri REL della commissione. Per la realizzazione dell’obiettivo sarà creato e condiviso un database informatico. Per quanto riguarda la risposta alla terapia innovativa ruxolitinib, la REL avvierà un’analisi dei pazienti trattati in real-life presso i centri referenti: sono in terapia 150 casi che potrebbero, secondo le stime d’incidenza della malattia, arrivare a circa 300 nello svolgimento del progetto. Per lo studio della risposta al trapianto di cellule staminali si utilizzerà un network nazionale-internazionale più ampio, come già utilizzato dal nostro gruppo in passato.
Disegno sperimentale dell’obiettivo #2.
La tecnologia NGS ha rivoluzionato lo studio delle malattie ematologiche permettendo il rapido sequenziamento di molti geni contemporaneamente e a costi inferiori rispetto all’approccio tradizionale tramite sequenziamento di Sanger. NGS sarà utilizzata per valutare lo stato mutazionale di pazienti affetti da NPM afferenti all’Unità di Ematologia di Varese e alle altre Unità della REL, qualora non disponibile l’analisi. Lo strumento che sarà utilizzato sarà il MiniSeqTM System (Illumina) e le analisi verranno eseguite presso l’Unità di Genetica Medica di Varese. Lo studio molecolare sarà eseguito partendo da campioni di sangue periferico dei pazienti e le diverse popolazioni cellulari verranno separate tramite gradiente di densità con Ficoll. Il DNA sarà estratto con kit commerciali (MiniKit – Qiagen) dai neutrofili (cellule del clone mieloide) e dai linfociti T (controllo costituzionale). I linfociti T verranno selezionati positivamente tramite biglie magnetiche coniugate con anticorpi anti-CD3 (Dynabeads CD3, Invitrogen). Il DNA controllo permetterà l’identificazione delle varianti somatiche rilevanti per la malattia. Il protocollo prevede l’utilizzo del pannello TruSight® Myeloid (Illumina) comprendente 54 geni frequentemente mutati nella neoplasie mieloidi. In particolare il pannello copre le intere regioni esoniche di 15 geni e regioni hotspots di mutazione di altri 39 geni. Di seguito la lista dei geni analizzati: ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC, CDKN2A, CEBPA, CSF3R, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1, JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1 e ZRSR2. Per l’analisi NGS è necessaria la preparazione delle librerie di sequenziamento: il protocollo TruSight® Myeloid utilizza 50 ng di DNA non frammentato e genera una libreria di 568 ampliconi di ~ 250 bp. Il workflow per la preparazione della libreria prevede l’ibridazione di più coppie di oligonucleotidi alle regioni da sequenziare, un processo di estensione e ligazione degli oligonucleotidi, l’amplificazione con PCR e incorporazione degli indici campione-specifici e la conversione a single strand della libreria. Le librarie verranno successivamente caricate sul MiniSeqTM System per la fase di sequenziamento (2 x 150bp). Utilizzeremo software dedicati per convertire i raw data files in files di analisi (FASTQ, BAM, VCF) per permettere l’allineamento delle sequenze ottenute al genoma di riferimento (hg19) e contro il genoma costituzionale dei singoli pazienti. Tale confronto permetterà l’identificazione delle varianti patogeniche somatiche. La presenza di tali varianti verrà confermata con sequenziamento di Sanger.
Disegno sperimentale dell’obiettivo #3
Questa fase del progetto è rivolta ai pazienti con TE e MF che non presentano le phenotype driver mutations, cioè che non presentano le mutazioni di JAK2, CALR, MPL. Sono circa l’8-10% della casistica di TE e MF ed abbiamo a disposizione circa 25 casi a Varese con MF. L’array CGH è una tecnica di citogenetica molecolare che permette di visualizzare, in un unico esperimento, microdelezioni/duplicazioni cromosomiche, traslocazioni sbilanciate e mosaicismi maggiori del 20%. Questa tecnica si basa sulla comparazione quantitativa di due campioni di DNA genomico: il DNA test e il DNA reference, marcati in maniera differente con due fluorocromi (Cy3 e Cy5), che vengono fatti ibridare contemporaneamente sulla superficie dell’array, dove si trovano sintetizzate sonde oligonucleotidiche. Questi cloni corrispondono a specifici loci di ogni singolo cromosoma e vanno a ricoprire l’intero genoma umano. Dal numero di cloni presenti dipende la risoluzione dell’array; la piattaforma da noi utilizzata comprende 180K oligonucleotidi, che consentono di avere una risoluzione media di 20kb. Una volta avvenuta l’ibridazione il risultato è dato dall’emissione delle due fluorescenze, le cui intensità vengono misurate dall’apposito scanner. In caso di un assetto cromosomico normale si avrà la medesima intensità di segnale tra i due DNA, mentre in presenza di delezioni/duplicazioni il segnale sarà sbilanciato, indicando la presenza di un’alterazione genomica. La tecnica array CGH prevede diverse fasi, di cui in dettaglio: estrazione con Maxi kit Quiagen (per l’estrazione del DNA dalle cellule nucleate del sangue/midollo, separate mediante Ficoll, si utilizza il Maxi kit della Quiagen, come da protocollo partendo da 6 cc di sangue periferico in EDTA, o 3-4 cc di sangue midollare in EDTA); precipitazione del DNA (il DNA viene fatto precipitare mediante Sodio Acetato 3M a pH 5.2, EtOH 100% al fine di migliorarne la qualità); quantificazione (il DNA test e il DNA reference – pool di DNA di individui sani – vengono quantificati utilizzando un nanofotometro Implein); marcatura (la concentrazione di DNA necessaria per l’analisi di Array CGH è di 1000 ng in un volume finale di 18 μl; I fluorocromi utilizzati sono CY3 per il Test e CY5 per il reference; i campioni vengono tenuti per 2 ore a 37°C, come da protocollo OGT); purificazione (la purificazione del prodotto di marcatura avviene mediante colonnine graduate Amicon Ultra 30K); denaturazione ed Ibridazione del campione su piattaforma 180K (i campioni vengono denaturati a 94°C 3’, 37°C 30’ e successivamente lasciati ibridare O.N. a 20 rpm, 65°C; la piattaforma utilizzata è a 180.000 oligonucleotidi (180K) con una risoluzione media di risoluzione di 20-30kb); lavaggi post-ibridazione (una volta dissemblata la camera di ibridazione, si allestiscono una serie di lavaggi in modo da eliminare i legami aspecifici e la fluorescenza in eccesso); acquisizione (L’array viene scansionato attraverso Microarray Scanner Innoscan 710); elaborazione dei dati (le immagini ottenute in seguito alla scansione vengono elaborate ed i dati analizzati con il software Cytosure) Si tenga conto che per ogni CNV riscontrata deve essere escluso che si tratti di polimorfismi noti con uso di databases internazionali. Qualora la CNV non sia mai stata riscontrata in letteratura dovrà essere effettuata la stessa analisi su campioni di controllo di linfociti T isolati dal paziente o sui genitori dei pazienti.
Disegno sperimentale dell’obiettivo #4
La definizione attuale della prognosi delle NMC si basa su criteri clinici. Nella MFP anche le mutazioni addizionali sembrano avere un ruolo prognostico. Nello svolgimento di questo obiettivo, un’analisi statistica ad hoc consentirà di generare dati per modelli prognostici anche tempo dipendenti integrando il dato clinico e quello molecolare. Le variabili continue saranno confrontate utilizzando il test non-parametrico Wilcoxon rank-sum. Le associazioni tra variabili categoriche (2-way tables) saranno testate con il Fisher exact test. L’incidenza cumulativa di trombosi, leucemia, mielofibrosi secondaria sarà stimata con l’analisi a rischi competitivi, considerando la morte come rischio competitivo. Il confronto tra incidenze sarà realizzato applicando il Pepe-Mori test, mentre l’effetto delle covariate continue con il modello di regressione di Fine-Gray. La sopravvivenza globale sarà stimata con Il metodo di Kaplan-Meier. L’analisi multivariata dei fattori predittivi di morte, trombosi, leucemia, risposta alla terapia con ruxolitinib e al trapianto di cellule staminali emopoietiche utilizzerà il modello di regressione di Cox. Il test Akaike sarà utilizzato per confrontare I nuovi modelli con quelli in uso per definirne la migliore efficienza.
Diagramma di Gantt
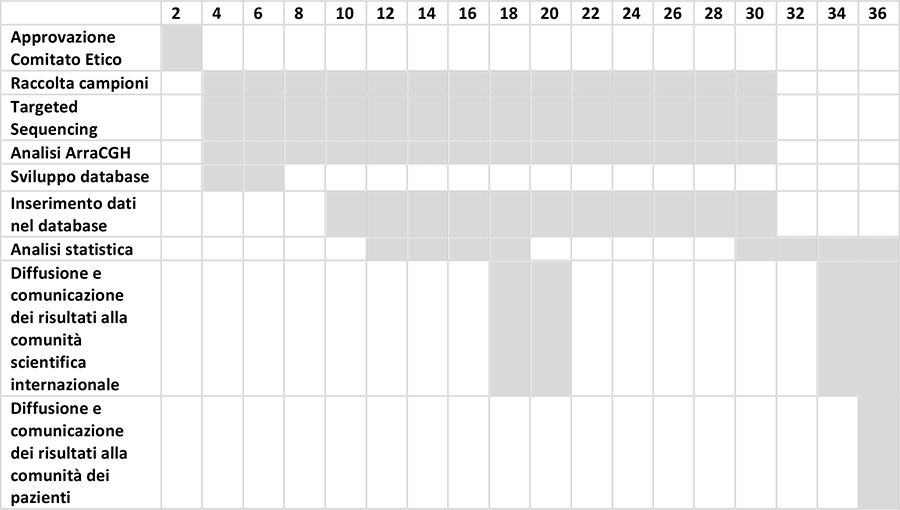
Risultati attesi
- Realizzazione di un database regionale delle NMP;
- Definizione della carta d’identità molecolare dei pazienti con NMC e di nuovi percorsi patogenetici nella TE e MF;
- Definizione di modelli prognostici per predire eventi e risposta alle terapie;
- Report annuale dei dati intermedi ottenuti dal progetto;
- Pubblicazione dei risultati su riviste scientifiche internazionali recensite e comunicazione ai gruppi pazienti con NMC.
Strumentazione a risorse disponibili
L’Unità di Ematologia consta di:
- Area degenza (12 letti)
- Area terapia cellulare (2 letti)
- Day-Hospital di ematologia (8 letti)
- Area ambulatoriale ematologica (5 sale)
- Trial Unit di ematologia
- Data Center di ematologia
L’Unità di Genetica consta dei seguenti strumenti:
- Sanger Automated Sequencer Abi Prism 410
- QFQ banding technique, cell cultures in Chang bone marrow/peripheral blood medium and analysis at fluorescence microscope
- Fluorescence microscope
- Thermocyclers, Real time PCR Rotor Q Quiagen and One step Plus Applied Biosystems technology and all instruments and technologies specific for cytogenetic and molecular genetic investigations (Nikon fluorescence microscope, cytogenetic analysis softwares, horizontal laminar air flow workstations, thermostats CO2, electrophoretic systems)
- Array-CGH technology (Technogenetics System) at 180K, for whole genome analysis.
- Analysis and interpretation Softwares: Technogenetics array-CGH software, Variant Studio from Illumina, and ISCA37, DECIPHER37 and DGV international database
- MiniSeqTM System (Illumina)
L’area informatica consta di:
- Microsoft Office Excel (Microsoft Corporation, Redmond, WA)
- R 3.1 (The R Project for Statistical Computing)
- Stata 11.2 (Stata- Corp, Lakeway, TX)
- Filemaker Pro
Significato e innovazione
Il significato di questo progetto di medicina personalizzata è incrementare la conoscenza delle NMC e definire lo stato mutazionale di pazienti. Quest’approccio servirà all’identificazione di categorie omogenee di pazienti. L’innovazione è l’impiego di tecnologie di NGS che in tempi rapidi consentono lo studio di molte alterazioni genomiche e la personalizzazione della cura. Infatti lo studio servirà ad assegnare ai pazienti la cura migliore al momento migliore della storia della malattia.
Bibliografia essenziale
- Passamonti F, Maffioli M, Caramazza D, Cazzola M. Myeloproliferative neoplasms: from JAK2 mutations discovery to JAK2 inhibitor therapies. Oncotarget, 2(6), 485-490 (2011).
- Mesa RA, Passamonti F. Individualizing Care for Patients With Myeloproliferative Neoplasms: Integrating Genetics, Evolving Therapies, and Patient-Specific Disease Burden. Am Soc Clin Oncol Educ Book, 35, e324-335 (2016).
- Passamonti F, Cervantes F, Vannucchi AM et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood, 115(9), 1703-1708 (2010).
- Passamonti F, Thiele J, Girodon F et al. A prognostic model to predict survival in 867 World Health Organization-defined essential thrombocythemia at diagnosis: a study by the International Working Group on Myelofibrosis Research and Treatment. Blood, 120(6), 1197-1201 (2012).
- Kroger N, Giorgino T, Scott BL et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood, 125(21), 3347-3350; quiz 3364 (2015).
- Passamonti F, Maffioli M, Cervantes F et al. Impact of ruxolitinib on the natural history of primary myelofibrosis: a comparison of the DIPSS and the COMFORT-2 cohorts. Blood, 123(12), 1833-1835 (2014).

